
Papers
[65] Deep learning for structure-based drug discovery
submitted
Jaechang Lim, Sang-Yeon Hwang, and Woo Youn Kim*
[64] Reversible Disorder-Order Transitions in Atomic Crystal Nucleation
submitted
Sungho Jeon, Taeyeong Heo, Sang-Yeon Hwang, Jim Ciston, Karen Bustillo, Bryan W. Reed, Jimin Ham, Sungsu Kang, Sungin Kim, Joowon Lim, Kitaek Lim, Ji Soo Kim, Min-Ho Kang, Ruth S. Bloom, Sukjoon Hong, Kwanpyo Kim, Alex Zettl, Woo Youn Kim, Peter Ercius*, Jungwon Park*, Won Chul Lee*
[63] PIGNet: A physics-informed deep learning model toward generalized drug-target interaction predictions
Seokhyun Moon,+ Wonho Zhung,+ Soojung Yang,+ Jaechang Lim and Woo Youn Kim∗
+equal contribution
[62] A holistic approach to the mechanism study of thermal degradation of OLED materials.
Accepted in J. Phys. Chem. A
Jin Woo Kim,+ Joonghyuk,+ Kim, Jaewook Kim, Jun Chwae, Sungwoo Kang, Soon Ok Jeon, Won-Joon Son, Hyeonho Choi, Byoungki Choi, Sunghan Kim, and Woo Youn Kim*
[61] ACE-Molecule: An open-source real-space quantum chemistry package
Sungwoo Kang+, Jeheon Woo+, Jaewook Kim, Hyeonsu Kim, Yongjun Kim, Jaechang Lim, Sunghwan Choi*, and Woo Youn Kim*
+equal contributions.
[60] Efficient construction of chemical reaction network guided by Monte Carlo tree search
Kyunghoon Lee+, Jin Woo Kim+, and Woo Youn Kim*
+equal contributions.
Chemical reaction networks are essential for the complete elucidation of chemical reaction mechanisms. Graph‐theoretic methods combined with quantum calculations are known to be an efficient approach with broad applicability for constructing reaction networks. However, this method entails high computational cost due to quantum calculations on chemically irrelevant intermediates coming from the exploration of a large scale chemical space. To remedy this problem, we propose to apply the Monte Carlo tree search algorithm to those graph‐theoretic methods. We have combined it with ACE‐Reaction, our in‐house graph‐theoretic method, and demonstrated its performance on five organic reactions. The result shows that the computational cost for quantum calculations has been reduced by up to 75 % from that of the original ACE‐Reaction. Furthermore, cost reduction became more efficient for more complex reactions.
화학반응 네트워크는 화학반응 메커니즘을 완전히 이해하는데 있어서 필수적이다. 양자 계산을 활용한 그래프 기반 방법론은 화학반응 네트워크를 만드는데 있어서 널리 효율적으로 활용할 수있는 방법이다. 그러나, 이 방법 역시 넓은 화학 공간을 탐색함에 따라 화학적으로 부적절한 중간체에 대하여 양자 계산을 하기 때문에 높은 계산 비용을 감수해야 한다. 이를 해결하기 위해, 우리는 MCTS을 그래프-기반 방법론에 적용할 것을 제안한다. 우리는 이를 우리가 예전에 개발한 ACE-Reaction에 적용해보고 그 성능을 평가하기 위해 5개의 유기반응에 대하여 적용해보았다. 그 결과 적용하기 전에 비해 최대 75%까지 양자 계산 비용을 줄일 수 있었다. 게다가, 그 효과는 복잡한 반응에 대하여 더 컸다.
[59] Graph theory-based reaction pathway searches and DFT calculations for the mechanism studies of free radical-initiated peptide sequencing mass spectrometry (FRIPS MS): a model gas-phase reaction of GGR tri-peptide
Jae-ung Lee+, Yeonjoon Kim+, Woo Youn Kim*, and Han Bin Oh*
+equal contributions.
Graph theory-based reaction pathway searches (ACE-Reaction program) and density functional theory (DFT) calculations were performed to shed light on the mechanisms for the production of [an+H]+, xn+, yn+, zn+, and [yn+2H]+ fragments formed in free radical-initiated peptide sequencing (FRIPS) mass spectrometry measurements of a small model system of glycine-glycine-arginine (GGR). In particular, the graph theory-based searches, which are rarely applied to gas-phase reaction studies, allowed us to investigate reaction mechanisms in an exhaustive manner without resorting to chemical intuition. As expected, radical-driven reaction pathways were favorable to charge-driven reaction pathways in terms of kinetics and thermodynamics. Charge- and radical-driven pathways for the formation of [yn+2H]+ fragments were carefully compared, and it was revealed that the [yn+2H]+ fragments observed in our FRIPS MS spectra originated from the radical-driven pathway, which is in contrast to the general expectation. The acquired understanding of the FRIPS fragmentation mechanism is expected to aid in the interpretation of FRIPS MS spectra. It should be emphasized that graph theory-based searches are powerful and effective methods for studying reaction mechanisms, including gas-phase reactions in mass spectrometry.
[58] Homochiral Supramolecular Thin Film from Self-Assembly of Achiral Triarylamine Molecules by Circularly Polarized Light.
Changjun Park, Jinhee Lee, Taehyoung Kim, Jaechang Lim, Jeyoung
Park, Woo Youn Kim, Sang Youl Kim *
[57] Molecular Generative Model Based On Adversarially Regularized Autoencoder
Seung Hwan Hong+, Jaechang Lim+, Seongok Ryu+, and Woo Youn Kim*
+equal contributions.
Deep generative models are attracting great attention as a new promising approach for molecular design. A variety of models reported so far are based on either a variational autoencoder (VAE) or a generative adversarial network (GAN), but they have limitations such as low validity and uniqueness. Here, we propose a new type of model based on an adversarially regularized autoencoder (ARAE). It basically uses latent variables like VAE, but the distribution of the latent variables is estimated by adversarial training like in GAN. The latter is intended to avoid both the insufficiently flexible approximation of posterior distribution in VAE and the difficulty in handling discrete variables in GAN. Our benchmark study showed that ARAE indeed outperformed conventional models in terms of validity, uniqueness, and novelty per generated molecule. We also demonstrated a successful conditional generation of drug-like molecules with ARAE for the control of both cases of single and multiple properties. As a potential real-world application, we could generate epidermal growth factor receptor inhibitors sharing the scaffolds of known active molecules while satisfying drug-like conditions simultaneously.

[56] Scaffold-based molecular design with a graph generative model
Jaechang Lim+, Sang-Yeon Hwang+, Seokhyun Moon, Seungsu Kim, and Woo Youn Kim*
+equal contributions.
Searching new molecules in areas like drug discovery often starts from the core structures of known molecules. The way as such has called for a strategy of designing derivative compounds retaining a particular scaffold as a substructure. On this account, our present work proposes a graph generative model that targets its use in scaffold-based molecular design. Our model accepts a molecular scaffold as input and extends it by sequentially adding atoms and bonds. The molecules generated are then guaranteed to contain the scaffold with certainty, and their properties can be controlled by conditioning the generation process on desired properties. The learned rule of extending molecules can well generalize to arbitrary kinds of scaffolds, including those unseen during learning. In the conditional generation of molecules, our model can simultaneously control multiple chemical properties, despite the search space constrained by fixed substructures. As a demonstration, we applied our model to designing inhibitors of the epidermal growth factor receptor and show that our model can employ a simple semi-supervised extension to broaden its applicability to situations where only a small amount of data is available.
새로운 분자를 찾는 과정은 종종 알려진 분자구조로 부터 출발한다. 이를 위해 본 연구에서는 scaffold 기반 그래프 분자 생성 모델을 개발하였다. 우리의 모델은 분자 scaffold를 input을 받고 이로 부터 원자와 화학결합을 추가하는 방식으로 그래프를 확장해 나아간다. 이를 통해 생성된 분자는 항상 주어진 scaffold를 substructure로 가지게 되며, 생성된 분자의 성질은 목표 물성에 대한 조건부 확률로서 조절될 수 있다. 이를 통해 학습된 분자 생성 규칙은 학습되지 않은 임의 scaffold에 대해서도 일반화 할 수 있었다. 조건부 분자 생성의 경우 분자 탐색 공간이 scaffold에 의해 제한됨에도 불구하고 여러 물성을 동시에 조절 할 수 있었다. 또한 개발된 모델이 epidermal growth factor receptor 저해제 개발과 같이 데이터의 양이 적은 경우에도 semi-supervised 방식으로 새로운 적용될 수 있음을 확인하였다.

[55] Deeply learning molecular structure-property relationships using attention- and gate-augmented graph convolutional network
[54] Predicting drug-target interaction using 3D structure-embedded graph representations from graph neural networks
Jaechang Lim, Seongok Ryu, Kyubyong Park, Yo Joong Choe, Jiyeon Ham, and Woo Youn Kim*
Accurate prediction of drug-target interaction (DTI) is essential for in silico drug design. For the purpose, we propose a novel approach for predicting DTI using a GNN that directly incorporates the 3D structure of a protein-ligand complex. We also apply a distance-aware graph attention algorithm with gate augmentation to increase the performance of our model. As a result, our model shows better performance than docking and other deep learning methods for both virtual screening and pose prediction. In addition, our model can reproduce the natural population distribution of active molecules and inactive molecules.
Drug-target interaction (DTI)를 정확하게 예측하는 것은 in silico drug design에 있어 매우 중요하다. 이를 위해 본 연구에서는 3차원 구조를 graph에 직접 포함시키는 graph nerual net 기반 DTI 예측방법을 개발하였다. 또한 3차원 거리기반 attention 알고리즘과 gate augmentation 방법을 적용하여 모델의 성능을 높였다. 결과적으로 제안된 모델은 기존의 docking이나 다른 deep learning 방법들에 비해서 virtual screening과 pose prediction 모두에서 보다 정확한 성능을 보였다 뿐만 아니라 제안된 모델은 active와 inactive 사이의 자연존재비율을 재현할 수 있었다.

[53] A Bayesian graph convolutional network for reliable prediction of molecular properties with uncertainty quantification
Seongok Ryu, Yongchan Kwon and Woo Youn Kim*
Deep neural networks have been increasingly used in various chemical fields. In the nature of data-driven approach, their performance strongly depends on data used in training. Therefore, models developed in data-hungry situations can cause highly uncertain predictions, leading to vulnerable decision making. Here, we show that Bayesian inference enables more reliable prediction with quantitative uncertainty analysis. Decomposition of the predictive uncertainty into model- and data-driven uncertainties allows us to elucidate the source of errors for further improvements. For molecular applications, we devised a Bayesian graph convolutional network (GCN) and evaluated its performance for molecular property predictions. Our study on the classification problem of bio-activity and toxicity shows that the confidence of prediction can be quantified in terms of the predictive uncertainty, leading to more accurate virtual screening of drug candidates than standard GCNs. The result of logP prediction illustrates that data noise affects the data-driven uncertainty more significantly than the model-driven one. On this hand, we could identify artefacts arisen from quantum mechanical calculations in the Harvard Clean Energy Project dataset. Consequently, the Bayesian GCN is critical for molecular applications under data-insufficient conditions.

[52] Performance of ACE-Reaction on 26 Organic Reactions for Fully Automated Reaction Network Construction and Microkinetic Analysis
Jin Woo Kim+, Yeonjoon Kim+, Kyung Yup Baek, Kyunghoon Lee, and Woo Youn Kim*
Accurate analysis of complex chemical reaction networks is necessary for reliable prediction of reaction mechanism. Though quantum chemical methods provide a desirable accuracy, large computational costs are unavoidable as considering numerous reaction pathways on the networks. We proposed a graph-theoretic approach combined with chemical heuristics (named ACE-Reaction) in previous work [ Chem. Sci. 2018, 9, 825], which automatically and rapidly finds out the most essential part of reaction networks just from reactants and products, and here we extended it by incorporating a stochastic approach for microkinetic modeling. To show its performance and broad applicability, we applied it to 26 organic reactions, which include 16 common functional groups. As a result, we could demonstrate that ACE-Reaction successfully found the accepted mechanism of all reactions, most within a few hours on a single workstation, and additional microkinetic modeling automatically discovered new competitive paths as well as a major path.

[51] Uncertainty quantification of molecular property prediction using Bayesian neural network models
Seongok Ryu, Yongchan Kwon, and Woo Youn Kim∗
Deep neural networks have outperformed existing machine learning models in various molecular applications. In practical applications, it is still difficult to make confident decisions because of the uncertainty in predictions arisen from insufficient quality and quantity of training data. Here, we show that Bayesian neural networks are useful to quantify the uncertainty of molecular property prediction with three numerical experiments. In particular, it enables us to decompose the predictive variance into the model- and data-driven uncertainties, which helps to elucidate the source of errors. In the logP predictions, we show that data noise affected the data-driven uncertainties more significantly than the model-driven ones. Based on this analysis, we were able to find unexpected errors in the Harvard Clean Energy Project dataset. Lastly, we show that the confidence of prediction is closely related to the predictive uncertainty by performing on bio-activity and toxicity classification problems.
[50] Single-crystalline Co2Si nanowires directly synthesized on silicon substrate for high-performance micro-supercapacitor
Jiyoung Lee, Chung-Yul Yoo, Yeong A Lee, Sang Hyun Park, Younghyun Cho, Jae Hyun Jun, Woo Youn Kim,* Bongsoo Kim,* and Hana Yoon*
[49] Quantum transport properties of single-crystalline Ag2Se0.5Te0.5 nanowires as a new topological material
Minjin Kim, Jihwan Kim, In-Ho Lee, Woo Hyun Han, Yun Chang Park, Woo Youn Kim,* Bongsoo Kim,* and Junho Suh*
[48] Poly(amide-imide) materials for transparent and flexible displays.
Sun Dal Kim, Byungyong Lee, Taejoon Byun, Jongmin Park, Isaac Shin, Namyoung Ahn, Myungeun Seo, Yunho Lee, Yeonjoon Kim, Woo Youn Kim, Hyukyun Kwon, Hanul Moon, Seunghyup Yoo, Sang Youl Kim*
Jaewook Kim, Sungwoo Kang, Jaechang Lim, and Woo Youn Kim*
Promising applications of graphdiyne have often been initiated by theoretical predictions especially using DFT known as the most powerful first-principles electronic structure calculation method. However, there is no systematic study on the reliability of DFT for the prediction of the electronic properties of the graphdiyne. Here, we performed a study of Li adsorption on the graphdiyne using hybrid DFT with LC-ωPBE and compared the results with those of PBE, because accurate prediction of the Li adsorption is important for performance as a Li storage that was first theoretically suggested and then experimentally realized. Our results show that PBE overestimates the adsorption energy inside a pore and the barrier height at the transition state of in-plane diffusion compared to the those of LC-ωPBE. In particular, LC-ωPBE predicted almost barrier-less in-plane diffusion of Li on the graphdiyne because of the presence of both in-plane and out-of-plane π orbitals. Also, LC-ωPBE favors a high spin state due to the exact exchange energy when several Li atoms are adsorbed on the graphdiyne, whereas PBE favors a low spin state. Thus, the use of the hybrid DFT is critical for reliable predictions on the electronic properties of the graphdiyne.
근래 주목받고 있는 탄소의 평면상 동소체 중 하나인 그래피다인(Graphdiyne)은 그 물리적, 화학적 특성이 이론적 방법, 특히 밀도범함수이론을 통하여 먼저 밝혀져 왔다. 그 예로 그래피다인은 그래핀보다 리튬 이온을 더 많이, 더 쉽게 흡착할 것으로 예측되었고, 실험 결과 리튬 이온 전지의 전극으로 활용 가능함이 밝혀졌다. 그러나 계산에 사용된 방법론의 신뢰도는 아직 제대로 점검된 적이 없었다. 본 연구에서는 순범함수인 PBE와 혼성범함수인 LC-ωPBE, 두 가지 방법을 사용하여 리튬의 흡착과 흡착된 리튬의 확산과정을 전산모사하였다. 그 결과 PBE는 LC-ωPBE에 비해 흡착 에너지 평면상 확산에 필요한 에너지를 과도하게 예측한것으로 확인되었다. 탄소-탄소 삼중결합에 있는 π 결합 오비탈들에 의해 리튬은 확산 과정에서 그래피다인 기질과의 결합을 거의 그대로 유지할 수 있었다. 또한 LC-ωPBE는 정확한 교환 에너지 항의 영향으로 PBE보다 스핀 상태를 더 높게 예측하였다. 이렇듯 혼성 범함수를 이용하는 것이 예측된 결과에 결과에 큰 영향을 끼칠수 있는것을 확인할 수 있었다.

[46] Molecular generative model based on conditional variational autoencoder for de novo molecular design.
Jaechang Lim, Seongok Ryu, Jin Woo Kim, and Woo Youn Kim*
We propose a molecular generative model based on the conditional variational autoencoder for de novo molecular design. It is specialized to control multiple molecular properties simultaneously by imposing them on a latent space. As a proof of concept, we demonstrate that it can be used to generate drug-like molecules with five target properties. We were also able to adjust a single property without changing the others and to manipulate it beyond the range of the dataset.
이 논문에서 de novo 분자 디자인을 위한 conditional variational autoencoder 기반 분자 생성모델을 제안한다. 이 모델은 여러가지 분자의 성질을 동시에 조절하는데 특화되어 있다. proof of concept으로 다섯가지 지정된 성질을 가지는 drug-like 분자를 생성하였다. 또한 나머지 성질을 고정시킨 상태에서 한 가지 성질만을 조절 할 수 있었으며, 데이터셋 범위 밖으로 한 가지 성질을 최적화 할 수 있었다.

Jaewook Kim, Sungwoo Kang, Jaechang Lim, Sang-Yeon Hwang, and Woo Youn Kim*
Real-space methods have not been suitable for hybrid density functional calculations due to high cost coming from the nonlocality of Fock operator. Here we propose a practical approach for fast computation. The key is to use a strictly local Kohn–Sham potential that can be deduced from any hybrid functional using the optimized effective potential method. This new approach improved the computation speed of the global and range-separated hybrid methods up to 30 and 80 times, respectively. As a result, accurate prediction of the size-dependent excitonic spectra of Si quantum dots became feasible.
본 논문에서는 유한차분법을이나 다중스펙트랄 방법과 같은 실공간 기저 함수를 사용하는 양자화학 프로그램에서 빠르게 혼성 범함수 계산을 수행할 수 있는 방법을 제안하였다. 최적화된 유효 포텐셜 방법 (OEP method)의 한 근사법인 Kriger-Li-Iafrate 방법론을 이용하여 교환 포텐셜을 나타내는 방법을 이용하여, 기존의 하트리-폭 연산자를 기반으로 한 혼성 범함수 계산에 비해 약 30~80배 빠르게 혼성 범함수 계산을 수행할 수 있었다. 이 방법을 이용 시간의존 밀도범함수이론(TDDFT) 계산을 수행하여 수 나노미터 크기의 실리콘 양자점 (Si QD) 계산도 수행할 수 있었다.

[44] Fragment-orbital tunneling currents and electronic couplings for analysis of molecular charge-transfer systems.
Sang-Yeon Hwang, Jaewook Kim, and Woo Youn Kim*
The tunneling current describes the flow of transferring electrons in charge transfer systems. It also enables one to compute the electronic coupling-matrix element, which is one of the most crucial quantity affecting the transfer rate. By using the frontier orbitals of molecular fragments and our own implementation of the theory, we showed the tunneling current gives coupling values with reasonable accuracy. Also, we showed how tunneling currents visualize the relation between structural variations and electron-flow patterns.
터널링 전류는 전하전달 분자 시스템에서 전자가 흐르는 모양을 보여주며, 또한 전달 속도를 결정하는 주요 물리량 중 하나인 전자 짝지음 상수를 계산할 수 있게 한다. 우리는 조각분자들의 HOMO/LUMO를 고유한 방법으로 이용하여, 터널링 전류가 합리적인 짝지음 상수들을 주는 것을 보였다. 또한, 시스템의 구조적 변화가 전하전달 패턴에 끼치는 영향을 터널링 전류가 어떻게 밝히는지도 보였다.

[43] Feasibility of activation energy prediction of gas-phase reactions via machine learning.
Sunghwan Choi, Yeonjoon Kim, Jin Woo Kim, Zeehyo Kim, and Woo Youn Kim*
We investigated the feasibility of machine learning models for the prediction of activation energies of gas-phase reactions. Three different models including the artificial neural network, the support vector regression, and the tree boosting methods were tested. We used the structural and thermodynamic properties of molecules and their differences as input features without resorting to specific reaction types so as to maintain the most general input form for broad applicability. The tree boosting method showed the best performance among others in terms of coefficient of determination, mean absolute error, and root-mean-square-error whose values were 0.89, 1.95 kcal/mol, and 4.49 kcal/mol, respectively. Computation time for the prediction of activation energies for 2,541 test reactions was about one second on a single computing node without using accelerators.
기체 상태 반응의 활성화에너지를 예측하기 위한 기계학습 모델을 개발하였고 모델의 정확도를 검증하였다. 인공신경망(ANN), 서포트 벡터 회귀분석(SVR), 트리 부스팅(TB)와 같은 모델을 만들고 활성화에너지 학습과 예측을 진행하였다. 특정 종류의 반응에 국한되지 않고 다양한 반응에 활용할 수 있는 모델을 만들기 위해, 반응 종류에 상관없이 구할 수 있는 반응물과 생성물의 구조적, 열역학적 성질을 포함한 값들을 입력 특성값으로 사용하였다. 테스트한 모델 중 TB 모델이 가장 정확도가 높았다 (R^2: 0.89, MAE: 1.95 kcal/mol, RMSE: 4.49 kcal/mol). 2541개의 테스트 반응에 대해 활성화에너지를 예측하는 데 불과 1초밖에 걸리지 않았다.

[42] On the achievement of high fidelity and scalability for large-scale diagonalizations in DFT simulations.
Sunghwan Choi, Woo Youn Kim, Min Sun Yeom, Hoon Ryu*
Recent advance in high performance computing (HPC) resources has opened the possibility to expand the scope of density functional theory (DFT) simulations toward large and complex molecular systems. This work proposes a numerically robust method that enables scalable diagonalizations of large DFT Hamiltonian matrices, particularly with thousands of computing CPUs (cores) that are usual these days in terms of sizes of HPC resources. The well‐known Lanczos method is extensively refactorized to overcome its weakness for evaluation of multiple degenerate eigenpairs that is the substance of DFT simulations, where a multilevel parallelization is adopted for scalable simulations in as many cores as possible. With solid benchmark tests for realistic molecular systems, the fidelity of our method are validated against the locally optimal block preconditioned conjugated gradient (LOBPCG) method that is widely used to simulate electronic structures. Our method may waste computing resources for simulations of molecules whose degeneracy cannot be reasonably estimated. But, compared to LOBPCG method, it is fairly excellent in perspectives of both speed and scalability, and particularly has remarkably less (< 10%) sensitivity of performance to the random nature of initial basis vectors. As a promising candidate for solving electronic structures of highly degenerate systems, the proposed method can make a meaningful contribution to migrating DFT simulations toward extremely large computing environments that normally have more than several tens of thousands of computing cores.

[41] Efficient structural elucidation of microhydrated biomolecules through interrogation of hydrogen bond networks.
Yeonjoon Kim, Jaewook Kim, Kyung Yup Baek,* and Woo Youn Kim*
There is no general way to elucidate stable hydrated structures even for simple amino acids because of the high complexity of chemical space. Here, we propose a very efficient computational method to selectively sample the most stable structures of microhydrated molecules. The key idea is to utilize the unique structural patterns of H-bond networks. As a proof of concept, we could identify the new global minima of glycine·10(H2O) and for the first time, we found the minimum number of water molecules required to stabilize the zwitterionic form of tyrosine. Furthermore, the most stable structures of hydrated glycine and tyrosine indeed had common features, which were consistent with the X-ray data of proteins in water.
생체 내 반응을 이해하기 위해서는 수화된 생분자의 안정한 구조를 찾아야 하지만 에너지면의 높은 복잡도 때문에 계산 비용이 많이 든다. 그렇기 때문에 에너지 극소점을 효율적으로 찾기 위한 새로운 방법을 개발하였다. 구조 샘플링 후 우리의 방법으로 각 구조의 수소 결합 네트워크를 분석해서 안정할 것으로 예상되는 후보 구조들을 빠르게 뽑을 수 있고, 이 구조들에 대해서만 양자 계산을 수행하기 때문에 계산량을 줄일 수 있다. 이 방법을 이용해서 물 분자 10개에 수화된 glycine의 가장 안정한 구조를 새로 제안하였고, tyrosine 쯔비터이온을 안정화시킬 수 있는 최소 물 분자 개수를 처음으로 찾아내었다. 그리고 찾아낸 안정한 구조가 실험에서의 X-ray 구조 분석 결과와도 일치함을 확인하였다.

Yeonjoon Kim, Jin Woo Kim, Zeehyo Kim, and Woo Youn Kim*
We propose a novel approach to rapidly search reaction paths in a fully automated fashion by combining chemical theory and heuristics. A key idea of our method is to extract a minimal reaction network composed of only favorable reaction pathways from the complex chemical space through molecular graph and reaction network analysis. It can be done very efficiently by exploring the routes connecting reactants and products with minimum dissociation and formation of bonds. Finally, the resulting minimal network is subjected to quantum chemical calculations to determine kinetically the most favorable reaction path at the predictable accuracy.
분자 그래프와 결합 네트워크 분석을 이용해 화학 반응 경로를 자동으로, 효율적으로 예측하는 방법을 개발하였다. 반응물로부터 생성물을 만들 때, 최소 개수의 결합이 생기고 끊어지는 경로들을 선호한다는 간단한 heuristic rule을 적용해서, 복잡한 chemical space로부터 minimal reaction network를 빠르게 추출해 낸다. 이 추출된 network에 기존의 양자계산 방법을 적용해 속도론적으로 선호되는 반응 경로를 찾을 수 있다.

Jaechang Lim, Sungwoo Kang, Jaewook Kim, Woo Youn Kim,* and Seol Ryu*
silver 나노입자의 plasmon 현상을 simlulation할 때, 큰 나노입자에서는 classical simulation이, 작은 나노입자에서는 TDDFT가 잘 작동한다. 반면에 TDDFT로 계산하기에는 비효율적이고 classical simulation을 하기에는 양자효과가 남아있는 10nm 정도 크기의 나노입자는 명확한 계산법이 없다. 이번 연구에서는 DDA 방법에서 bulk의 polarizability를 원자의 polarizability로 대체하는 ADA라는 방법을 제안하였다. 그 결과 작은 은 나노 입자의 스펙트럼 계산에서는 TDDFT와 정성적으로 유사한 결과를 얻었고, 기존의 DDA가 설명하지 못하는 LSPR peak의 blue shift를 재현할 수 있었다.

Jaewook Kim+, Kwangwoo Hong+, Sang-Yeon Hwang, Seongok Ryu, Sunghwan Choi, and Woo Youn Kim*
+equal contributions.
혼성 밀도 범함수(hybrid density functional) 방법을 이용할 때, 주로 하트리-폭 교환 연산자가 포함된 일반화된 콘-샴 식을 사용한다. 본 논문에서는 최적화된 유효 포텐셜 (optimized effective potential) 이론을 이용, 혼성 밀도 범함수 이론에 사용되어지는 콘-샴 포텐셜을 국소화 된 함수로 나타내는것이 여기상태 계산에 더 바람직하다는 것을 보여주었다. KLI(Krieger-Li-Iafrate) 근사를 이용한 혼성 포텐셜을 사용할 경우, 일반적인 방법에서와 달리 가상 오비탈이 N-전자 계의 성질을 보여주어 전기적으로 의해 들뜬 상태를 잘 표현하는 것을 알 수 있었다. 이 방법을 이용 시간의존 밀도범함수이론(TDDFT) 계산을 수행한 결과 LDA(국소밀도근사) 수준의 kernel만 가지고도 기존의 범함수들보다 정확하게 들뜬 상태 에너지를 얻을 수 있었다.

Jaechang Lim, Sunghwan Choi, Jaewook Kim, and Woo Youn Kim*
이 논몬에서는 real space grid method에 exact exchage Kohn-Sham orbital 기반 configuration interaction singles and doubles (CISD) 를 implement했다. 그 결과 기존의 Hartree-Fock orbital을 사용한 CISD에 비해서 작은 분자의 excitation energy를 계산하는데 있어서 더 적은 계산량으로도 더 정확한 결과를 얻을 수 있었다.

Sunghwan Choi*, Oh-kyoung Kwon, Jaewook Kim, and Woo Youn Kim*
They investigate the performance of heterogeneous computing using graphics processing units and many integrated cores with 20 CPU cores. By comparing Hartree potential evaluations for silver nanoparticles of various sizes, they found that grid-based electronic structure calculations could be maximally accelerated using a heterogeneous computer architecture.

Jaechang Lim, Sunghwan Choi, Sungwoo Kang, Jaewook Kim, Kwangwoo Hong, and Woo Youn Kim*
이 논문에서는 real space grid method기반의 initial guess를 개발하였다. Grid Cutting이라고 이름지은 이 방법은 작은 simulation box안에서 SCF를 통해서 initial guess를 만들어내게 된다. 결과적으로 기존의 Linear combination of atomic orbital, Superposition of atomic density, Huckel 방법들에 비해서 계산시간과 SCF cycle의 숫자를 줄일 수 있었다.

Minjune Kim+, Yena Kim+, Yeonjoon Kim+, Yongmin Kwon, Kyungrok Ham, Woo Youn Kim*, and Sang Woo Han*
+equal contributions.
We report a systematic study on the correlation of the modified electronic structure of nanocrystal catalysts with the adsorption properties of the substrate and the resultant catalytic activity by using Baeyer–Villiger oxidation catalyzed by composition-controlled Pt-based nanocubes (NCs) as a model heterogeneous catalysis reaction. The incorporation of 3d transition metals into Pt to form PtM (M=Zn, Co, and Ni) alloy NCs allowed fine-tuning of the electronic structure of Pt. PtM NCs with a higher-lying d band center exhibited higher catalytic performance owing to the enhanced initial activation of the carbonyl group of the substrate. This work emphasizes the importance of fine-tuning the electronic structure of heterogeneous catalysts to advance their catalytic function.

Sungwoo Kang, Seongok Ryu, Sunghwan Choi, Jaewook Kim, Kwangwoo Hong, and Woo Youn Kim*
The projector augmented wave (PAW) method was implemented in a quantum chemistry package that uses Lagrange-sinc basis set, namely ACE-Molecule. Its numerical accuracy has been assessed for the AE6 test set and compared with that of Hartwigsen-Goedecker-Hutter type pseudopotentials with nonlinear core correction. The PAW method shows a rapid convergence toward complete basis set limits of all electron calculations, whereas the pseudopotential method has a significant deviation even at small grid spacing. To alleviate spurious egg-box effects, the so-called supersampling method is adopted to the operation of projector functions. It improved accuracy of total energy calculations at substantially large grid spacing but did not show significant difference in atomization energies from the results without supersampling, which is due to error cancellation.
Seongok Ryu, Sunghwan Choi, Kwangwoo Hong, and Woo Youn Kim*
The egg-box effect, the spurious variation of energy and force due to the discretization of continuous space, is an inherent vexing problem in grid-based electronic structure calculations. Its effective suppression allowing for large grid spacing is thus crucial for accurate and efficient computations. We here report that the supersampling method drastically alleviates it by eliminating the rapidly varying part of a target function along both radial and angular directions. In particular, the use of the sinc filtering functionperforms best because as an ideal low pass filter it clearly cuts out the high frequency region beyond allowed by a given grid spacing.
Kyungjun Kang, Jaewook Kim, Ansoo Lee, Woo Youn Kim, and Hyunwoo Kim*
A mild and efficient dehydrative cross-coupling reaction between allylic alcohols and N-heterocycles using palladium catalysis is reported. A bicyclic bridgehead phosphoramidite (briphos) ligand together with Pd(dba)2 is a highly efficient catalyst, and an acid additive involved in the rate-determining step promotes the catalytic cycle. The coupling reaction of allylic alcohols with N-heterocycles including imidazoles, benzimidazoles, and triazole proceeds under mild reaction conditions with high yields using Pd/briphos and pentafluorophenol.

Hahn Kim, Yeonjoon Kim, Jaewook Kim, and Woo Youn Kim*
We devised a useful computational strategy to search for new types of stable two-dimensional carbon allotropes. As an illustration, we considered all the isomers of C18H6 and found three stable building units for graphynes. Then, we investigated the stability of each unit {hexagonal/triangular/parallelogrammatic C18H6 (C18H6h/C18H6t/C18H6p)} in that π-conjugation effects improve the stability and conductivity of the system. The resonance energies of the above C18H6 are smaller than that of benzene, but still large; among the three types, C18H6t is the most stable, followed by C18H6. This supports why the graphyne composed of C18H6t was already synthesized, while it is likely that the graphynes composed of C18H6h and C18H6p could also be synthesized. The graphynes composed of C18H6h and C18H6p were predicted to have zero band gaps with Dirac cones, whereas that composed of C18H6t turned out to be a semiconductor with the band gap of 0.49 eV. In particular, C18H6p has not only direction-dependent Dirac cones but also their unconventional locations in the first Brillouin zone.

Jaewook Kim+, Kwangwoo Hong+, Sunghwan Choi, Sang-Yeon Hwang, and Woo Youn Kim*
We developed a program code of configuration interaction singles (CIS) based on a numerical grid method. We used Kohn–Sham (KS) as well as Hartree–Fock (HF) orbitals as a reference configuration and Lagrange-sinc functions as a basis set. Our calculations show that KS-CIS is more cost-effective and more accurate than HF-CIS. The former is due to the fact that the non-local HF exchange potential greatly reduces the sparsity of the Hamiltonian matrix in grid-based methods. The latter is because the energy gaps between KS occupied and virtual orbitals are already closer to vertical excitation energies and thus KS-CIS needs small corrections, whereas HF results in much larger energy gaps and more diffuse virtual orbitals. KS-CIS using the Lagrange-sinc basis set also shows a better or a similar accuracy to smaller orbital space compared to the standard HF-CIS using Gaussian basis sets. In particular, KS orbitals from an exact exchange potential by the Krieger–Li–Iafrate approximation lead to more accurate excitation energies than those from conventional (semi-) local exchange–correlation potentials.

Yeonjoon Kim and Woo Youn Kim*
We present a powerful method for the conversion of molecular structures from atomic connectivity to bond orders to three-dimensional (3D) geometries. There are a number of bond orders and 3D geometries corresponding to a given atomic connectivity. To uniquely determine an energetically more favorable one among them, we use general chemical rules without invoking any empirical parameter, which makes our method valid for any organic molecule. Specifically, we first assign a proper bond order to each atomic pair in the atomic connectivity so as to maximize their sum and the result is converted to a SMILES notation using graph theory. The corresponding 3D geometry is then obtained using force field or ab initio calculations. This method successfully reproduced the bond order matrices and 3D geometries of 10000 molecules randomly sampled from the PubChem database with high success rates of near 100% except a few exceptional cases. As an application, we demonstrate that it can be used to search for molecular isomers efficiently.

Sunghwan Choi+, Kwangwoo Hong+, Jaewook Kim, and Woo Youn Kim*
+equal contributions.
We developed a self-consistent field program based on Kohn-Sham density functional theory using Lagrange-sinc functions as a basis set and examined its numerical accuracy for atoms and molecules through comparison with the results of Gaussian basis sets. The result of the Kohn-Sham inversion formula from the Lagrange-sinc basis set manifests that the pseudopotential method is essential for cost-effective calculations. The Lagrange-sinc basis set shows faster convergence of the kinetic and correlation energies of benzene as its size increases than the finite difference method does, though both share the same uniform grid. Using a scaling factor smaller than or equal to 0.226 bohr and pseudopotentials with nonlinear core correction, its accuracy for the atomization energies of the G2-1 set is comparable to all-electron complete basis set limits (mean absolute deviation ≤1 kcal/mol). The same basis set also shows small mean absolute deviations in the ionization energies, electron affinities, and static polarizabilities of atoms in the G2-1 set. In particular, the Lagrange-sinc basis set shows high accuracy with rapid convergence in describing density or orbital changes by an external electric field. Moreover, the Lagrange-sinc basis set can readily improve its accuracy toward a complete basis set limit by simply decreasing the scaling factor regardless of systems.
Jaewook Kim, Kwangwoo Hong, Sunghwan Choi, and Woo Youn Kim*
This paper is dedicated to Professor Kwan Kim on the occasion of his honorable retirement.
We report feature of Kohn-Sham (KS) molecular orbitals computed with the Krieger-Li-Iafrate (KLI) approximation for exact exchange through the comparison to the results of Hartree-Fock (HF) and other KS methods such as local density approximation (LDA) and generalized gradient approximation (GGA). KLI occupied orbitals have similar energies and shapes with those of HF. KLI virtual orbitals are likely to form bound states with negative eigenvalues due to the correct -1/r asymptotic behavior of KLI potentials, whereas HF virtual orbitals are mostly unbound. As a result, HF orbitals tend to be diffuse because of their plane-wave-like nature, and their energies are highly sensitive to the size of basis set. The energies of LDA/GGA orbitals appear to be upshifted by a constant factor from the KLI results, but they also produce unbound virtual orbitals like HF. The energy gaps between KLI occupied and virtual orbitals are very close to the corresponding experimental excitation energies compared to the other methods. We also show that Brillouin's theorem can be applied to a Slater determinant made of KLI orbitals as a corollary of the KLI approximation.
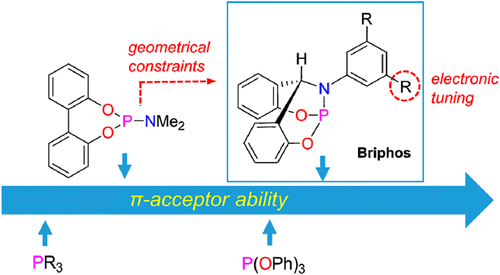
Ansoo Lee, Seihwan Ahn, Kyungjun Kang, Min-Seob Seo, Yeonjoon Kim, Woo Youn Kim*, and Hyunwoo Kim*
A new type of bicyclic bridgehead phosphoramidites (briphos) is reported, where the geometrical constraints significantly enhance the π-acceptor ability compared with its monocyclic analogs. The briphos is shown to be highly efficient and tunable for Rh(I)-catalyzed conjugate additions of aryl boronic acids to α,β-unsaturated ketones and N-tosyl ketimines.
Minjune Kim+, Yeonjoon Kim+, Jong Wook Hong+, Seihwan Ahn, Woo Youn Kim*, and Sang Woo Han*
+equal contributions.
A systematic study of heterogeneous Buchwald–Hartwig amination using shape-controlled Pd nanocrystals with distinctly different surface facets is presented.
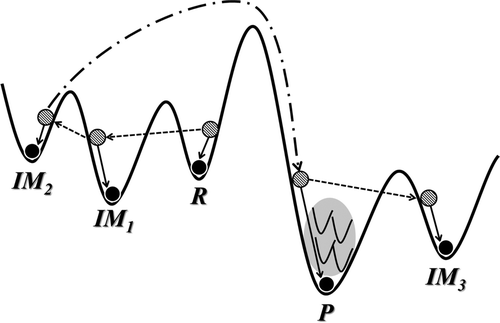
Yeonjoon Kim, Sunghwan Choi, and Woo Youn Kim*
Basin-hopping sampling has been widely used for searching local minima on a potential energy surface. Reaction intermediates including reactants and products are also local minima composed of a reaction path, but their brute-force sampling is too demanding because of large degrees of freedom. We developed an efficient Monte Carlo basin-hopping method to sample reaction intermediates through the fragmentation of molecules and a post-analysis scheme using the graph theory with a matrix representation of molecular structures. The former greatly reduces the dimension of a given potential energy surface, while the latter offers not only the effective screening of resulting local minima toward desirable intermediates but also their automatic ordering along a reaction path. We combined it with the density functional tight binding method for rapid calculations and tested its performance for organic reactions.
Yeonchoo Cho, Seung Kyu Min, Jeonghun Yun, Woo Youn Kim, Alexandre Tkatchenko, and Kwang S. Kim*
The complexes of a DNA base bound to graphitic systems are studied. Considering naphthalene as the simplest graphitic system, DNA base–naphthalene complexes are scrutinized at high levels of ab initio theory including coupled cluster theory with singles, doubles, and perturbative triples excitations [CCSD(T)] at the complete basis set (CBS) limit. The stacked configurations are the most stable, where the CCSD(T)/CBS binding energies of guanine, adenine, thymine, and cytosine are 9.31, 8.48, 8.53, 7.30 kcal/mol, respectively. The energy components are investigated using symmetry-adapted perturbation theory based on density functional theory including the dispersion energy. We compared the CCSD(T)/CBS results with several density functional methods applicable to periodic systems. Considering accuracy and availability, the optB86b nonlocal functional and the Tkatchenko–Scheffler functional are used to study the binding energies of nucleobases on graphene. The predicted values are 18–24 kcal/mol, though many-body effects on screening and energy need to be further considered.

Kwangwoo Hong and Woo Youn Kim*
What's your spin on it? A carbon nanotube device decorated with single-molecule magnets through π–π stacking interactions (see scheme) was shown to have spin-dependent transmission (red=spin-up, blue=spin-down) around the energy levels of the molecule owing to Fano resonance, which could be tuned by adjusting the strength of the π–π stacking. This Fano-resonance-driven spin-valve effect offers a new method to make molecular spintronics.

Seonki Hong, Yun Suk Na, Sunghwan Choi, In Taek Song, Woo Youn Kim,* and Haeshin Lee*
Polydopamine is the first adhesive polymer that can functionalize surfaces made of virtually all material chemistries. The material-independent surface modification properties of polydopamine allow the functionalization of various types of medical and energy devices. However, the mechanism of dopamine polymerization has not yet been clearly demonstrated. Covalent oxidative polymerization via 5,6-dihydroxyindole (DHI), which is similar to the mechanism for synthetic melanin synthesis, has been the clue. Here, it is reported that a physical, self-assembled trimer of (dopamine)2/DHI exists in polydopamine, which has been known to be formed only by covalent polymerization. It is also found that the trimeric complex is tightly entrapped within polydopamine and barely escapes from the polydopamine complex. The result explains the previously reported in vitro and in vivo biocompatibility. The study reveals a different perspective of polydopamine formation, where it forms in part by the self-assembly of dopamine and DHI, providing a new clue toward understanding the structures of catecholamines such as melanin.
Sang-Il Choi, Su-Un Lee, Woo Youn Kim,* Ran Choi, Kwangwoo Hong, Ki Min Nam,Sang Woo Han,* and Joon T. Park*
Modification of the electronic structure and lattice contraction of Pt alloy nanocatalysts through control over their morphology and composition has been a crucial issue for improving their electrocatalytic oxygen reduction reaction (ORR) activity. In the present work, we synthesized PtCo alloy nanocubes with controlled compositions (PtxCo NCs, x = 2, 3, 5, 7, and 9) by regulating the ratio of surfactants and the amount of Co precursor to elucidate the effect of the composition of nanocatalysts on their ORR activity. PtxCo NCs had a Pt-skin structure after electrochemical treatment. The electrocatalysis experiments revealed a strong correlation between ORR activity and Co composition. Pt3Co NCs exhibited the best ORR performance among the various PtxCo NCs. From density functional theory calculations, a typical volcano-type relationship was established between ORR activity and oxygen binding energy (EOB) on NC surfaces, which showed that Pt3Co NCs had the optimal EOB to achieve the maximum ORR activity. X-ray photoelectron spectroscopy and X-ray diffraction measurements demonstrated that the electronic structure and lattice contraction of the PtxCo NCs could be tuned by controlling the composition of NCs, which are highly correlated with the trends of EOB change.

[18] Chromium Porphyrin Arrays as Spintronic Devices
J. Am. Soc. Chem. 133, 9364-9369 (2011) .
Woo Jong Cho, Yeonchoo Cho, Seung Kyu Min, Woo Youn Kim, and Kwang S. Kim*
[17] The origin of dips for the graphene-based DNA sequencing device.
Phys. Chem. Chem. Phys. 13, 14293-14296 (2011).
Yeonchoo Cho, Seung Kyu Min, Woo Youn Kim, and Kwang S. Kim*
[16] Fast DNA sequencing with a graphene-based nanochannel device.
Nature Nanotech. 6, 162-165, (2011).
Seung Kyu Min+, Woo Youn Kim+, Yeonchoo Cho, and Kwang S. Kim*
+equal contributions.
[15] Radical Polymer as 2-Dimensional Organic Half Metal.
Chem. Eur. J. 16, 12141, (2010).
E. C. Lee, Y. C. Choi, Woo Youn Kim, N. J. Singh, S. Lee, J. H. Shim, & Kwang S. Kim
[14] Tuning Molecular Orbitals in Molecular Electronics and Spintronics.
Acc. Chem. Res. 43, 111 (2010)
Woo Youn Kim & Kwang S. Kim
[13] Near-field focusing and magnification through self-assembled nanoscale spherical lenses.
Nature 460, 498 (2009)
Ju Young Lee, Byung Hee Hong, Woo Youn Kim, Seung Kyu Min, Yukyung Kim, Mikhail V. Jouravlev, Ranojoy Bose, Keun Soo Kim, In-Chul Hwang, Laura J. Kaufman, Chee Wei Wong, Philip Kim & Kwang S. Kim
[12] Effect of electrodes on electronic transport of molecular electronic devices.
J. Phys. Chem. A 113, 4100 (2009).
Yeon Choo Cho, Woo Youn Kim* & Kwang S. Kim*
*co-corresponding author
[11] Neutral and Anionic Gold Decamers: Planar Structure with Unusual Spatial Charge-Spin Separation.
J. Chem. Theory Comput. 5, 1216 (2009).
Young Cheol Choi, Woo Youn Kim, Han Myoung Lee & Kwang S. Kim
[10] Application of quantum chemistry in nanotechnology: spin and electron transport phenomena through molecular junctions.
Chem. Soc. Rev. 38, 2319 (2009).
Woo Youn Kim, Young Cheol Choi, Seung Kyu Min, Yeon Choo Cho & Kwang S. Kim
[9] Understanding structures and electronic/spintronic properties of single molecules, nanowires, nanotubes, and nanoribbons towards the design of nanodevices.
J. Mater. Chem. 18, 4510 (2008).
Woo Youn Kim, Young Cheol Choi & Kwang S. Kim
[8] Prediction of very large values of magnetoresistance in a graphene nanoribbon device.
Nature Nanotech. 3, 408 (2008).
Woo Youn Kim & Kwang S. Kim
[7] Carbon Nanotube, Graphene, Nanowire, and Molecule-Based Electron and Spin Transport Phenomena Using the Nonequilibrium Green's Function Method at the Level of First Principles Theory.
J. Comput. Chem. 29, 1073 (2008).
Woo Youn Kim & Kwang S. Kim
[6] Negative differential resistance of carbon nanotube electrodes with asymmetric coupling phenomena.
Phys. Rev. B 76, 033415 (2007).
Woo Youn Kim, S. K. Kwon & Kwang S. Kim.
[5] How can we make stable single atomic linear chains? gold-cesium binary subnanowires as an example of charge-transfer-driven alloying approach.
Phys. Rev. Lett. 98, 076101 (2007).
Young Cheol Choi, Han Myoung Lee, Woo Youn Kim, S. K. Kwon, T. Nautiyal, D.-Y. Cheng, K. Vishwanathan & Kwang S. Kim
[4] Magic structures and quantum conductance of [110] silver nanowires.
Phys. Rev. Lett. 96, 096104 (2006).
Dayong Cheng, Woo Youn Kim,* Seung Kyu Min, Tashi Nautiyal & Kwang S. Kim*
*co-corresponding author
[3] Anomalous behavior of mercury in one-dimension: Density functional calculations.
Phys. Rev. B 71, 113104 (2005).
Woo Youn Kim, Tashi Nautiyal, Suk Joo Youn & Kwang S. Kim
[2] Role of molecular orbitals of the benzene in electronic nanodevices.
J. Chem. Phys. 122, 094706 (2005).
Young Cheol Choi, Woo Youn Kim, Kee-Su Park, P. Tarakeshwar, Kwang S. Kim, Tae-Suk Kim & Jin Yong Lee
[1] Insights into the structures, energetics, and vibrations of monovalent cation-(water)1-6 clusters.
J. Phys. Chem. A 108, 2949 (2004).
Han Myoung Lee, P. Tarakeshwar, Jungwon Park, Maciej Roman Kołaski, Yeo Jin Yoon, Hai-Bo Yi, Woo Youn Kim & Kwang S. Kim
BOOKS, CHAPTERS
Computer Aided Nanomaterials Design – Self-assembly, Nanooptics, Molecular Electronics/Spintronics, and Fast DNA Sequencing
Yeonchoo Cho, Seung Kyu Min, Ju Young Lee, Woo Youn Kim, and Kwang S Kim

Molecular Spintronics
"Computational Methods for Large Systems: Electronic Structure Approaches for Biotechnology and Nanotechnology", edited by Jeffery R. Reimers, John Wiley & Sons, Inc., Hoboken, NJ, USA. 2011.
Woo Youn Kim & Kwang S. Kim